Understanding Quality Control in Pharma and Biopharma: The Pillars of Product Integrity
What is Quality Control in Pharma and Biopharma?
Quality Control (QC) in the pharmaceutical and biopharmaceutical industries is a systematic process designed to verify that drugs, biologics, and medical products meet predefined standards of identity, strength, quality, purity, and potency. It encompasses a series of tests, inspections, and analyses performed throughout the manufacturing lifecycle—from raw material receipt to final product release—to ensure consistency, safety, and efficacy. QC operates under stringent regulatory frameworks like those from the FDA, EMA, and ICH guidelines, emphasizing Good Manufacturing Practices (GMP) to prevent defects, contamination, or deviations that could harm patients.
Unlike Quality Assurance (QA), which focuses on preventive measures and process design, QC is reactive and detective, providing empirical evidence that products are compliant. In biopharma, where products like vaccines and monoclonal antibodies involve living cells and complex biomolecules, QC is even more vital due to inherent variability in biological processes. Ultimately, QC bridges the gap between R&D innovation and market-ready therapies, fostering trust in the healthcare supply chain.

The Role of QC in the Pharma and Biopharma Industries
In the traditional pharmaceutical sector, QC ensures small-molecule drugs (e.g., generics and oral solids) are free from impurities and stable over time, supporting high-volume production and cost efficiency. For biopharma, which handles large-molecule biologics (e.g., insulin, gene therapies), QC mitigates risks from upstream fermentation or downstream purification, where microbial or viral contaminants could amplify exponentially.
Key contributions include:
- Patient Safety: Detecting adulterants or instabilities that could lead to adverse events, as evidenced by past recalls like contaminated heparin in 2008.
- Regulatory Approval: Generating data for IND/NDA submissions and post-market surveillance, ensuring compliance with cGMP and ISO standards.
- Cost Savings: Early identification of batch failures reduces waste, rework, and litigation—critical in biopharma where a single batch can cost millions.
- Global Harmonization: Aligning with pharmacopeias (USP, EP, JP) to facilitate international trade and supply chain resilience.
As the industry shifts toward personalized medicines and continuous manufacturing, QC evolves with technologies like real-time analytics and AI-driven predictions, maintaining its role as the industry’s quality gatekeeper.
Sub-Departments in QC: Specialized Guardians of Quality
QC is typically organized into specialized sub-departments, each focusing on distinct aspects of product testing. These units collaborate to provide a holistic quality profile, with tests often overlapping for comprehensive validation. Below, we elaborate on key sub-departments, their responsibilities, and core tests.
QC Analytical: The Backbone of Chemical and Physical Characterization
QC Analytical handles the physicochemical analysis of raw materials, in-process samples, and finished products, ensuring they match specifications for composition, purity, and performance. This department uses instrumental techniques to quantify active pharmaceutical ingredients (APIs), excipients, and degradation products, crucial for both pharma (e.g., tablet dissolution) and biopharma (e.g., protein folding in antibodies).
Core tests and elaborations:
- High-Performance Liquid Chromatography (HPLC): Separates and quantifies compounds in complex mixtures, detecting impurities at ppm levels per ICH Q3A/B guidelines. In biopharma, it’s adapted for glycan profiling in glycoproteins.
- Dissolution and Disintegration Testing: Measures drug release rates in simulated body fluids (USP <711>), vital for bioavailability in oral solids—failure here flags formulation issues.
- Spectroscopic Methods (UV-Vis, FTIR, NMR): Identifies molecular structures and monitors stability, with FTIR being non-destructive for real-time process control.
- Particle Size Analysis (e.g., Laser Diffraction): Ensures uniform distribution in injectables or suspensions, preventing dosing inaccuracies.
This department’s data supports stability studies (ICH Q1A) and release decisions, with automation enhancing throughput in high-volume pharma settings.

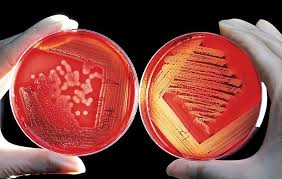
QC Microbiology: Defending Against Microbial Threats
QC Microbiology focuses on preventing and detecting microbial contamination, as detailed in prior discussions. It tests for bacteria, fungi, and endotoxins across non-sterile and sterile products, emphasizing environmental controls in GMP facilities.
Core tests and elaborations:
- Microbial Limits Testing (USP <61>/<62>): Enumerates total aerobes, yeasts, and molds in non-sterile products; specified pathogens like E. coli are absent in pharma oral drugs.
- Environmental Monitoring: Active air sampling and surface swabbing in cleanrooms (ISO 14644), tracking colony-forming units (CFUs) to validate aseptic processing.
- Sterility Testing (USP <71>): Membrane filtration or direct inoculation for injectables, incubating samples for 14 days to confirm zero growth—essential for biopharma vaccines.
- Bacterial Endotoxin Test (BET, USP <85>): LAL-based assays detect pyrogens at <5 EU/mg, critical for parenteral products to avoid febrile reactions.
In biopharma, where cell cultures are vulnerable, this department integrates rapid PCR methods for faster detection, reducing hold times.
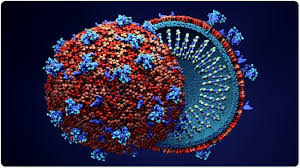
QC Virology: Safeguarding Against Viral Contaminants
QC Virology is a specialized arm, particularly prominent in biopharma, tasked with ensuring viral safety in products derived from biological sources (e.g., cell lines, animal tissues). It detects adventitious viruses that could hitchhike through production, using sensitive assays to comply with ICH Q5A guidelines for viral clearance validation.
Core tests and elaborations:
- Viral Plaque Assays: Quantifies infectious viruses by counting plaques on cell monolayers, used for retrovirus detection in monoclonal antibody production.
- qPCR and NAT (Nucleic Acid Testing): Amplifies viral genomes for early detection of DNA/RNA viruses like parvovirus or HIV, with limits of <10 copies/mL in biopharma intermediates.
- In Vivo Viral Safety Tests: Inoculate animals (e.g., mice) with product to observe symptoms, though increasingly supplemented by in vitro alternatives like TCID50 (tissue culture infectious dose).
- Viral Clearance Studies: Validate inactivation/removal steps (e.g., nanofiltration) using model viruses like MMV (minute virus of mice), ensuring >4 log reduction.
This department is indispensable for biologics like blood products or gene therapies, where viral risks could lead to transmission outbreaks, and it collaborates with regulatory bodies for risk-based assessments.
Animal House: Conducting Potency and Safety Tests In Vivo for Veterinary Vaccines
The Animal House (or vivarium) sub-department performs in vivo studies to evaluate veterinary vaccine potency (efficacy) and safety (toxicity), adhering to GLP (Good Laboratory Practice) and 3Rs principles (Replacement, Reduction, Refinement). It’s integral for pre-release validation of each vaccine batch, ensuring market readiness by confirming the product’s ability to protect target species without causing harm. In veterinary biopharma, human-relevant models are adapted to livestock like cows, testing immunogenicity, pharmacokinetics, and real-world safety before batch approval.
Core tests and elaborations:
- Potency Assays: Measure biological activity by injecting a representative sample from the vaccine batch into target animals (e.g., cows) and assessing antibody titers after 28 days via serological methods like ELISA or virus neutralization tests. This ensures the batch elicits the intended immune response at specified doses, meeting pharmacopeial standards (e.g., OIE or USDA guidelines) for protective efficacy against diseases like foot-and-mouth or bovine respiratory syncytial virus.
- Safety Studies (Adverse Reaction Observation): Administer the full vaccine batch dose to sentinel animals (e.g., groups of cows) and monitor for local/systemic adverse reactions over 7-14 days, including injection site swelling, fever, lethargy, or anaphylaxis. These acute observations flag batch-specific risks like hypersensitivity, integrating genotoxicity checks (e.g., Ames test) and ensuring no dose-limiting toxicities before market release.
- Pharmacodynamic Models: Disease-specific challenge tests, such as vaccinating cows followed by controlled pathogen exposure to quantify protection (e.g., reduced clinical signs or viral shedding), confirming the therapeutic index and batch-to-batch consistency.
Ethical oversight via IACUC (Institutional Animal Care and Use Committee) ensures humane practices, with a push toward organoids, in vitro alternatives, and AI models to minimize animal use while maintaining rigorous pre-release safety and potency data for veterinary vaccine batches.minimize animal use. In pharma, it’s key for generics bioequivalence; in biopharma, for IND-enabling data.

Integrated Key Tests and Future Outlook
Across these sub-departments, overarching tests like stability (accelerated/real-time per ICH Q1) and raw material ID (FTIR/HPLC) ensure end-to-end quality. Integration via LIMS (Laboratory Information Management Systems) streamlines data flow, enabling holistic batch review.
As pharma/biopharma advances with mRNA tech and ATMPs (advanced therapy medicinal products), QC sub-departments will leverage PAT (Process Analytical Technology) for predictive quality. This evolution underscores QC’s timeless mission: delivering safe, effective therapies that transform lives.